
Girl With Peculiar Rash and Developmental Delay

History
A 12-year-old girl presents with a “peculiar” rash and developmental delay. The rash was noted soon after birth and was initially vesicular; the vesicles subsequently resolved. Around the child’s first birthday, the mother noticed dark streaks on her back and extremities that have persisted (A). Recently, she noticed whitish streaks on the child’s arms and legs (B).
The child has had intermittent eye crossing since 1 year of age and failed vision testing at age 12 years. An ophthalmologist who evaluated the child diagnosed Duane syndrome type 1 in the left eye and bilateral
myopic astigmatism.
She has had frequent urinary tract infections since age 1 year. Two voiding cystourethrograms at ages 2 years and 6 years revealed a neurogenic bladder with no ureteral reflux. She requires intermittent catheterization.
In addition to significant developmental delay, the child has a history of seizure disorder, scoliosis (requiring bracing at night), chronic constipation, and multiple contractures.
Her surgical history includes removal of a hydrocolpos at age 3 days; resection of dermoid tumors in nasofrontal and lumbar locations at age 2 years; and hamstring and Achilles tendon releases at ages 3 years and 11 years, respectively. She has also had 2 tethered cord release surgeries for her neurogenic bladder.

Physical examination
The well-developed, nourished girl has normal vital signs. She is alert, active, and speaks in complete sentences. Pulmonary, cardiac and abdominal findings are unremarkable.
HEENT: Her head is normocephalic with normally formed ears. Her vision is 20/80 bilaterally, which improves to 20/50 with eyeglasses. Intraocular pressures are normal to finger palpation. Motility testing reveals restriction of abduction of the left eye, with mild narrowing of the lid fissure (C). There is mild lid fissure narrowing on abduction. Fundus findings are normal. An oral examination reveals many areas of tooth decay and numerous fillings (D). Her hair is wiry and sparse (E).

Skin: She has flat areas of hypopigmentation and hyperpigmentation in a V-shaped pattern on her back and a swirled pattern on the anterior aspect of her trunk. Atrophic, hypopigmented linear streaks are present on her extremities.
Back: Moderate scoliosis is evident, with a 40-degree upper right thoracic curve and a 30-degree left lower lumbar curve.
Extremities: She has bilateral Achilles contractures, with inability to flex the foot to 90 degrees, and shortening of the phalanges and nail ridging (F).
In addition, she has some atrophy of the dorsal hand muscles.
Neurological: She has global hypertonia with resistance to passive and active movement. Reflexes are 21 in all extremities. Sensation is globally intact. She toe-walks with a limp. Cranial nerves appear grossly intact.
TO WHAT DIAGNOSIS DO THESE FINDINGS POINT—AND WHAT IS THE PROGNOSIS?
(Answers and discussion on next page.)
ANSWERS:
The girl has incontinentia pigmenti (IP), an uncommon, X-linked dominant neurocutaneous disorder characterized mainly by dermatologic manifestations, classically pigmentation along Blaschko lines. The condition, also known as Bloch-Sulzberger disease, is caused by mutations in the IKBKG gene (previously known as NEMO), located on chromosome Xq28. It affects girls almost exclusively because it is fatal in boys.
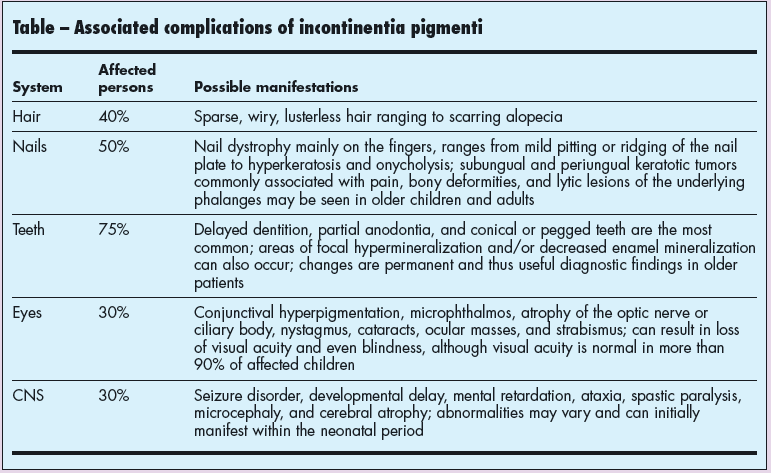
Although skin manifestations may be the sole presentation for IP, more than 80% of affected children have other associated defects (Table). Ophthalmic and neurological defects can potentially lead to blindness, mental retardation, and seizures; however, serious complications are rare. Genetic counseling is indicated for affected persons.
DERMATOLOGIC MANIFESTATIONS
Lyonization of the X chromosome creates functional mosaicism, which manifests as areas of normal skin coloration from active normal X chromosomes intermixed with areas of abnormal skin differentiation from inactivated X chromosomes. This results in the characteristic whorled pattern of pigmentation of classic IP. The cutaneous expression of IP generally occurs in 4 stages, all of which may not be present in a given patient.
The vesicular stage is generally present at birth or develops within the first 2 weeks of life. It consists of red papules and/or vesicles on an erythematous base that follow Blaschko lines. The lesions develop predominantly on the extremities but may also occur circumferentially on the trunk and rarely on the head or neck. Resolution generally occurs by 4 months of age, although recurrence during febrile illnesses of childhood is possible.
The verrucous stage manifests as thickened, warty-appearing linear and/or whorled plaques on an erythematous base in a linear pattern. The lesions occur most commonly on affected limbs and rarely on the face or trunk. They involute spontaneously within 6 months of appearance.
The hyperpigmented (pigmentary) stage, or classic IP, is characterized by macular streaks and/or whorls of brown or slate-gray pigmentation that follow Blaschko lines. The lesions are mainly distributed on the trunk, although their location does not necessarily correlate with areas of prior skin involvement during the earlier vesicular and verrucous stages. They commonly develop within the first few weeks of life (sometimes at birth), may overlap previous stages, and generally resolve slowly by adolescence.
The hypopigmented (atrophic) stage is distinguished by hairless, hypopigmented, atrophic reticulated or linear patches found mainly on the lower extremities, specifically the flexor aspect of the lower legs. The arms and trunk are less commonly affected. The lesions are a late manifestation of IP; they generally develop during adolescence and persist into adulthood.
RECOGNITION AND REFERRAL
IP is typically diagnosed solely on clinical grounds through physical examination and family history. Genetic testing may be offered to patients in cases with nonclassic features but is expensive. About 80% of affected persons have a deletion in the IKBKG gene. In addition, genetic testing may be used to identify asymptomatic
female carriers of the gene if the genetic mutation in the family is known.
MANAGEMENT AND PROGNOSIS
Treatment and further diagnostic studies are variable and should be tailored to the individual patient based on the presence—and absence—of associated conditions.
This patient’s seizures and constipation were well-controlled with medication. She is currently undergoing a trial of Botox injections to her Achilles tendon along with physiotherapy for recurrence of contractures. A Z-lengthening procedure for the tendon may be considered if this therapy is unsuccessful. She is scheduled to undergo a Xiao procedure for her neurogenic bladder in the near future. The importance of continuing regular neurological, ophthalmologic, dental, orthopedic, and urologic examinations was stressed to the mother.
The patient and her family need to be counseled about the risk of transmitting the genetic defect to her offspring. The child has about a 33% risk of each of the following: having a normal female child, having a normal male child, having an affected female child, and having a miscarriage of an affected male child. The patient could also be offered preimplantation testing of the embryo by using in vitro fertilization if she desires to have children but does not want to risk miscarriage. n
FOR MORE INFORMATION:
Barbagallo JS, Kolodzieh MS, Silverberg NB, Weinberg JM. Neurocutaneous disorders. Dermatol Clin. 2002;20(3):547-560, viii.
Hadj-Rabia S, Froidevaux D, Bodak N, et al. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003;139(9):1163-1170.
Meuwissen ME, Mancini GM. Neurological findings in incontinentia pigmenti; a review. Eur J Med Genet. 2012;55(5):323-331.