
Kaposiform Hemangioendo-thelioma Complicated by Kasabach-Merritt Phenomenon
Laura Pruitt, BS
Brody School of Medicine at East Carolina University, Greenville, North Carolina
Amy L. Gagnon, MD, and Barbara B. Wilson, MD
University of Virginia School of Medicine, Charlottesville, Virginia
An infant girl was noted to have jaundice with edema and violaceous discoloration of her right forearm at birth. She had been born via uncomplicated vaginal delivery at 36 weeks of gestation, and the discoloration initially was attributed to the umbilical cord having been wrapped around her arm. Over the next 3 weeks, however, the edema and discoloration increased.
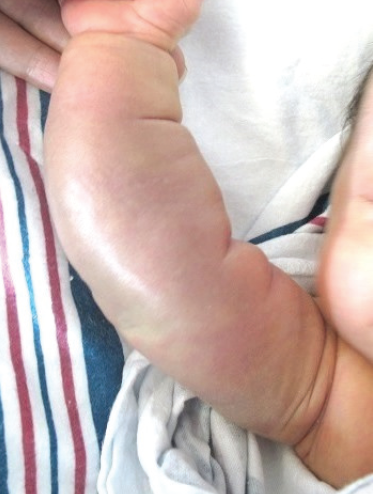 Physical examination findings were significant for a firm, edematous, dusky, ill-defined mass involving the right arm. Ultrasonography revealed a high-flow vascular lesion, and the neonate was admitted to the hospital for magnetic resonance imaging (MRI) of her right forearm and initiation of propranolol for a presumed infantile hemangioma. MRI scans revealed infiltrative soft tissue within the musculature and subcutaneous fat of the right forearm, and lymphatic malformations in the right upper extremity.
Physical examination findings were significant for a firm, edematous, dusky, ill-defined mass involving the right arm. Ultrasonography revealed a high-flow vascular lesion, and the neonate was admitted to the hospital for magnetic resonance imaging (MRI) of her right forearm and initiation of propranolol for a presumed infantile hemangioma. MRI scans revealed infiltrative soft tissue within the musculature and subcutaneous fat of the right forearm, and lymphatic malformations in the right upper extremity.
Results of blood work at admission demonstrated a platelet count less than 10,000/µL, anemia, an elevated D-dimer level, hypofibrinogenemia, and persistent hyperbilirubinemia.
Based on the patient’s history, examination findings, and diagnostic test results, a diagnosis of kaposiform hemangioendothelioma (KHE) complicated by Kasabach-Merritt phenomenon (KMP) was made.
Weekly intravenous infusions of vincristine at 0.05 mg/kg were added to her treatment regimen of propranolol at 2 mg/kg, 3 times a day. Over the next 4 weeks, the patient was readmitted to the hospital twice for lethargy and acute swelling of her right forearm. The patient’s medication was switched from vincristine to sirolimus 0.2 mL orally twice a day.
FIgure: The neonate’s firm, edematous, dusky mass with ill-defined borders on the forearm, characteristic of KHE.
Discussion
KHE is a rare, locally aggressive, vascular tumor named for its infiltrative nature and its histologic characteristics that are similar to Kaposi sarcoma.1 KHE can occur at any age but most commonly occurs in infancy and childhood, without predilection for sex.2 KHE may occur anywhere on the body but is found most commonly on the extremities as a violaceous, indurated plaque with ill-defined borders. KHE also commonly occurs on the trunk, neck, and face, and in the retroperitoneum. The lesions can be painful.
The diagnosis of KHE typically is made based on clinical findings and results of imaging studies. Biopsy often is avoided due to the risk of bleeding.1 The most common presentation is a rapid enlargement of the lesion and cutaneous discoloration. These soft tissue tumors are enhanced on MRI scans with contrast and show ill-defined margins, involvement of multiple tissue planes, overlying cutaneous thickening, edema, subcutaneous stranding, and signal voids secondary to hemosiderin deposits.3
KHE has a mortality rate of 10% to 37% due to direct tumor invasion and KMP.4 KMP is characterized by a consumptive coagulopathy with severe, persistent thrombocytopenia, microangiopathic anemia, hypofibrinogenemia, and an elevated D-dimer level.
Two tumors are associated with KMP: KHE and tufted angioma. These tumors often are described as being on a spectrum of disease, with tufted angioma being a superficial form of KHE, although whether these tumors are separate entities is controversial.2 Unlike true infantile hemangiomas, KHE and tufted angiomas are present at birth and have both vascular and lymphatic components, with small, convoluted capillaries that favor turbulent blood flow and consequent platelet activation and intravascular coagulation.1-3 Platelet transfusion should be avoided because it can lead to tumor enlargement and worsening coagulopathy.1
KHE shows little tendency to spontaneously involute, and treatment often is difficult. Various treatment options exist, but none have been uniformly effective. Surgical excision and arterial embolization have limited roles due to the infiltrative nature of KHE, the associated coagulopathy, and the technical difficulty of cannulating very small feeder vessels.2,5
Systemic corticosteroids, interferon alfa, chemotherapeutic agents (including vincristine and cyclophosphamide), ticlopidine, and aspirin have been used alone and in combination with variable responses. Successful treatment with propranolol monotherapy has been reported in less-extensive lesions.4 Vincristine has been used alone and in combination with other therapies and has reported response rates of 86% to 100%, but it has not been used widely because of its risk of neurotoxicity and other potential adverse effects.2,4,5
A recent study of the use of sirolimus in the treatment of KHE demonstrated a 100% response rate.6 The average response time was 5.3 days, and the average stabilization time of platelets was 15.1 days. Adverse effects were minimal and reversible, and neither dose adjustment nor discontinuation of the treatment was necessary. Given the risk of immunosuppression, trimethoprim-sulfamethoxazole also was started for Pneumocystis carinii pneumonia prophylaxis.6
The classic case of KHE discussed here demonstrates the importance of distinguishing KHE from infantile hemangiomas, which are not associated with KMP and often regress spontaneously,3 and suggests that sirolimus is a promising new treatment for patients with refractory KHE.
References
1. Wang Z, Li K, Yao W, Dong K, Xiao X, Zheng S. Steroid-resistant kaposiform hemangioendothelioma: a retrospective study of 37 patients treated with vincristine and long-term follow-up [published online ahead of print October 24, 2014]. Pediatr Blood Cancer. doi:10.1002/pbc.25296.
2. Fahrtash F, McCahon E, Arbuckle S. Successful treatment of kaposiform hemangioendothelioma and tufted angioma with vincristine. J Pediatr Hematol Oncol. 2010;32(6):506-510.
3. Croteau SE, Liang MG, Kozakewich HP, et al. Kaposiform hemangioendothelioma: atypical features and risks of Kasabach-Merritt phenomenon in 107 referrals. J Pediatr. 2013;162(1):142-147.
4. Choeyprasert W, Natesirinilkul R, Charoenkwan P. Successful treatment of mild pediatric Kasabach-Merritt phenomenon with propranolol monotherapy. Case Rep Hematol. 2014;2014:364693.
5. Hermans DJJ, van Beynum IM, van der Vijver RJ, Kool JSL, de Blaauw I, van der Vleuten CJM. Kaposiform hemangioendothelioma with Kasabach-Merritt syndrome: a new indication for propranolol treatment. J Pediatr Hematol Oncol. 2011;33(4):e171-e173.
6. Kai L, Wang Z, Yao W, Dong K, Xiao X. Sirolimus, a promising treatment for refractory kaposiform hemangioendothelioma. J Cancer Res Clin Oncol. 2014;140(3):471-476.